방한한 헬릭스미스 인허가본부장 “엔젠시스 美 시판 허가 준비 착수...FDA와 논의 계속”
-
기사 스크랩
-
공유
-
댓글
-
클린뷰
-
프린트

투 도안 헬릭스미스 인허가본부장은 최근 기자와 만나 “미국 식품의약국(FDA)과 논의하며 엔젠시스의 신약 허가 신청에 필요한 데이터를 정리하고 있다”며 이같이 말했다. 지난 6월 미국에서 헬릭스미스에 합류한 투 본부장은 엔젠시스 규제와 관련된 현안을 본사 연구진과 직접 논의하기 위해 지난 15일 한국을 방문했다.
규제업무(RA)는 인력난에 허덕이는 바이오 업계에서 특히 인재 유치가 어려운 분야로 꼽힌다. RA는 신약 임상과 상용화 과정에서 규제당국과 소통하는 업무를 아우르는 분야다. 임상에서 아무리 긍정적인 데이터를 확보하더라도 RA 담당자가 규제당국의 눈높이를 맞출 수 있는 데이터를 갈무리 하지 못하면 신약 상용화는 성공할 수 없다. 연구개발, 투약, 치료 후 관리 등에 골고루 이해도가 높으면서 미국에서 신약을 내놓는 데 관여한 RA 전문가를 국내 벤처기업이 확보하는 건 ‘하늘에 별 따기’라는 게 업계 이야기다.
투 본부장은 FDA와 논의를 거쳐 신약 허가를 받아 본 경험이 있는 RA 전문가다. 그는 미국 카이트파마에서 세계 최초 CAR-T(키메릭 항원수용체 T세포) 치료제인 예스카타의 RA 업무를 담당했다. 지난 15년간 미국 바이오젠아이덱, 앨러간 등에서도 근무하며 역량을 쌓았다. 헬릭스미스에서는 유전자치료제 엔젠시스의 신약 허가 신청 시 FDA에 제출해야 할 항목들이 무엇인지를 검토하고 이와 관련한 데이터를 사내에서 확보하는 게 그의 일이다.
투 본부장은 입사 후 임상에서 유효성을 확보하는 데에 초점을 맞추고 있는 사내 분위기를 바꾸는 데 공을 들였다. 당뇨변성신경병증(DPN)을 대상으로 한 엔젠시스의 두 번째 임상 3상 결과가 내년 나오는 만큼 임상과 별도로 신약 허가를 신청하기 위한 사전 작업에 착수해야 할 때라는 판단에서였다.
투 본부장은 “임상 3상은 신약의 유효성을 입증하는 것도 중요하지만 시판 허가 신청에 쓸 서류를 규제당국의 요구에 맞춰 마련하는 ‘레이블링’ 작업도 필요하다”며 “FDA가 어떠한 데이터를 요구하는지를 확인하고 신약허가신청서에 담을 레이블링 항목들을 채우는 작업을 하고 있다”고 말했다.
엔젠시스를 개발하는 헬릭스미스도 CAR-T 치료제를 개발한 카이트파마처럼 ‘최초’에 도전하고 있다. 아직 엔젠시스와 같은 플라스미드DNA 치료제로 미국에서 시판허가를 신청한 사례는 없다. 투 본부장은 “최초라고 해서 기존 신약의 RA 과정과 다른 부분은 없다”며 “항체치료제든, 유전자치료제든 신약과 관련된 정보들을 규제당국에 투명하게 공개하며 FDA와 신뢰를 쌓는 게 무엇보다 중요하다”고 말했다.
규제당국의 요구 중 충족하기 어려운 부분에 대해서도 끊임없이 규제당국과 소통해 대안을 모색해야 한다는 게 투 본부장의 설명이다. 그는 “FDA를 규제기관이 아닌 자문기관으로서 바라볼 필요도 있다”며 “과학과 데이터를 기반으로 한 주장을 제시하면 FDA는 열린 자세로 기업들의 질의 사항에 대응해주고 있다”고 말했다.
투 본부장은 향후 헬릭스미스의 자회사인 카텍셀이 개발 중인 CAR-T 치료제의 RA 업무도 맡을 예정이다. 카텍셀은 2023년 CAR-T 치료제로 임상 1상을 진입할 계획이다.
이주현 기자 deep@hankyung.com
관련 뉴스
-
1
SK바이오팜은 자체 개발한 뇌전증 신약 ‘세노바메이트’(미국 제품명 엑스코프리)를 글로벌 헬스케어 기업 엔도그룹에 기술수출했다고 23일 밝혔다. 캐나다 시장 출시를 위한 것으로, 계약금은 200...
-
2
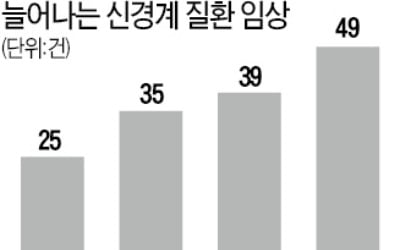
세계적으로 고령 인구가 빠르게 증가하면서 알츠하이머 치매와 파킨슨병 등 퇴행성 신경 질환을 치료하려는 시도가 크게 늘고 있다. 고령 인구 증가 추이를 고려할 때 신경 질환 치료제 시장이 항암제 못지않은 거대 시장으로...
-
3
증상 발현 후 20일이 넘은 코로나19 확진자를 일반병실로 옮기라는 정부 명령을 놓고 의료현장에서 “현실을 제대로 감안하지 않은 탁상행정”이라는 비판 목소리가 커지고 있다. 중환자실 입원 환자가...

![K팝 업계에도 '친환경' 바람…폐기물 되는 앨범은 '골칫거리' [연계소문]](https://img.hankyung.com/photo/202206/99.27464274.3.jpg)


