"中 임상만으론 FDA 승인 어렵다"…美 제약업계로 번진 중국 리스크
-
기사 스크랩
-
공유
-
댓글
-
클린뷰
-
프린트
10일(현지시간) 로이터통신 등에 따르면 FDA 외부 자문위원회는 일라이릴리와 이노벤트바이오로직스가 함께 개발한 폐암 치료신약 '티비트'가 FDA 허가를 받기 위해선 추가 데이터가 필요하다고 결정했다. 이날 자문위 투표에 참여한 15명 중 14명이 당장 허가하기엔 임상 결과가 부족하다는 데 표를 던진 것으로 알려졌다.
FDA는 다음달 말께 티비트 허가 여부를 최종 결정할 계획이다. FDA가 자문위 권고를 의무적으로 따라야 하는 것은 아니지만 대개 자문위와 같은 판단을 내린다. 티비트는 물론 중국에서 임상 시험을 하고 있는 다른 의약품의 미국 진출에 차질이 빚어질 것이란 전망이 나오는 이유다.
일라이 릴리는 중국 바이오기업인 이노벤트와 함께 티비트를 개발했다. 이 후보물질은 중국에서 진행한 임상시험을 통해 무진행생존기간(PFS) 등 신약 허가를 위한 주요 목표치를 달성했다. 이날 자문위 회의에 참여한 위원들도 이 치료제가 암 진행이나 사망을 늦추는 데 효과 있다는 점은 인정한 것으로 알려졌다.
FDA는 이번 자문위 결정이 다른 의약품 허가에도 영향을 미칠 것으로 내다봤다. 중국에서 개발을 진행 중인 치료제 후보물질 중 FDA 허가를 앞둔 제품은 25개에 이른다.
FDA의 신약 허가 분위기가 달라졌다는 분석도 나온다. 중국에서 임상시험을 주로 진행했던 베이진의 림프종 치료제 브루킨사가 2019년 FDA 허가를 받았기 때문이다. 당시 파듀 국장은 품질 데이터가 있다면 중국에서 진행한 임상시험 결과도 받아들일 수 있다고 밝혔다.
릴리와 노바티스 등은 값싼 중국 의약품을 미국에 들여오는 전략을 추진해왔다. 중국 바이오기업들도 약값이 저렴한 자국보다 해외 진출을 선호했다. 이런 제약사들의 신약 개발 전략에도 제동이 걸릴 것이란 전망이 나온다.
이에 대해 파듀 국장은 "비인두암처럼 미국보다 아시아에 많은 질환이라면 중국 단일 임상 데이터를 더 유연하게 받아들일 수 있을 것"이라고 했다. 개발하는 의약품의 적응증이나 국가별 질환 특성 등에 따라 평가 결과가 달라질 수 있다는 것이다.
이지현 기자 bluesky@hankyung.com
-
1
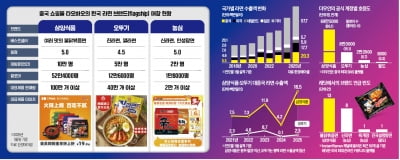
삼양식품의 ‘불닭볶음면’을 앞세운 K라면이 일시적 유행을 넘어 세계 최대 라면 소비국의 일상에 파고들고 있다. 대(對)중국 월간 수출액은 올해 1월 기준 처음 2000만달러(약 290억원)를 넘...
-
2
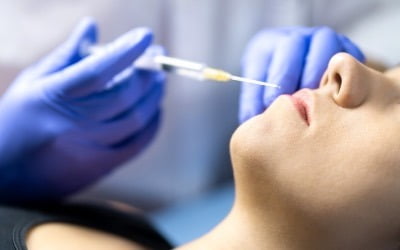
"주사 한 방에 아기 피부 된다"…톱스타들 사이 '난리'
코스닥 상장사 파마리서치의 의료기기 브랜드 ‘리쥬란(rejuran)’의 글로벌 구글 검색량이 최근 스위스 IBSA의 ‘프로필로(profhilo)’를 앞질렀다. 인플루언서와의...
-
3
 [마켓칼럼]불안정하지만 결국은 증시 상승…韓 시장에도 기회 있어
[마켓칼럼]불안정하지만 결국은 증시 상승…韓 시장에도 기회 있어
※한경 마켓PRO 텔레그램을 구독하시면 프리미엄 투자 콘텐츠를 보다 편리하게 볼 수 있습니다. 텔레그렘에서 ‘마켓PRO’를 검색하면 가입할 수 있습니다.김영민 토러스자산운용 대표이사트럼프 정책의...

![K팝 업계에도 '친환경' 바람…폐기물 되는 앨범은 '골칫거리' [연계소문]](https://img.hankyung.com/photo/202206/99.27464274.3.jpg)


