프리시젼바이오, 자회사 코로나19 항원진단제품 美 정식 승인
-
기사 스크랩
-
공유
-
댓글
-
클린뷰
-
프린트
나노디텍은 2021년 12월 미국 식품의약국(FDA)으로부터 코로나19 진단 제품에 대한 긴급사용승인(EUA)을 받았다. 이후 지난해 3월 FDA가 발표한 EUA 제품의 전환 계획과 같은 해 5월 코로나19 공중보건비상사태(PHE) 종료에 맞춰, 제공된 가이드라인에 따라 허가용 임상시험을 추가로 진행하며 510(k)를 준비해 왔다.
FDA는 전환 계획에 따라 총 19종의 제품을 정식승인(510(k))했다. 이번에 승인된 나노디텍의 ‘Nano-Check COVID-19 Antigen Test’는 20번째 승인 제품이며, 육안으로 진단하는 전문가용 신속진단으로는 최초의 승인 제품이라고 회사는 설명했다.
김한신 프리시젼바이오 대표는 “대유행 종료에도 불구하고 코로나19는 인플루엔자 제품과 유사하게 일상적인 계절성 전염병으로 진화하면서 지속적으로 진단 수요가 발생하고 있다”며 “미국에서 정식 승인을 받아 현지 생산시설을 보유한 나노디텍에서 제품을 공급하게 된다면 EUA 종료 이후 미국 시장을 선점하는 효과가 있을 것”이라고 말했다. 이어 “이를 바탕으로 북중남미 시장에서의 영향력도 높일 수 있을 것으로 기대한다”고 했다.
한편 나노디텍은 지난해부터 호흡기 세포융합 바이러스(RSV) 진단 제품 및 코로나19와 인플루엔자 감염 여부를 동시에 검사하는 콤보 제품 등의 미국 임상을 진행하고 있다. 임상이 마무리 되는대로 오는 3월 FDA 510(k)를 신청할 예정이다.
김예나 기자 yena@hankyung.com
-
1
메디웨일, '심장질환 예측' AI 기기 세브란스에 공급
의료 인공지능(AI) 기업 메디웨일은 연세대 세브란스병원에 ‘닥터눈(Reti-CVD)’을 공급하고 이달부터 안과에서 처방이 시작된다고 23일 밝혔다.닥터눈은 망막 촬영을 통해 심장 컴퓨터단층촬영...
-
2
암 환자는 방사선 치료를 받는 1~2개월간 한여름에도 목욕을 하지 못한다. 사인펜 등으로 표시한 기준선이 지워지지 않도록 유지해야 해서다. 서울대병원 방사선종양학과 교수로 매일 환자를 진료하는 우홍균 파프리카랩 대표...

-
3
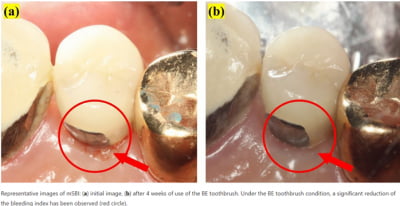
프록시헬스케어, 네이처에 "생체전류 칫솔 사용시 임플란트 환자 출혈지수 개선" 연구결과 게재
프록시헬스케어가 개발한 트로마츠 생체전류 칫솔이 임플란트 환자 출혈지수 개선 효과를 입증했다. 해당 논문은 지난 12월 4일 세계 3대 학술지 네이처 저널에 게재됐다. 국내 한해 임플란트 시술 건수는 50만...

![K팝 업계에도 '친환경' 바람…폐기물 되는 앨범은 '골칫거리' [연계소문]](https://img.hankyung.com/photo/202206/99.27464274.3.jpg)


